


 Lou Gehrigova choroba (amyotrofická laterální skleróza ALS, též Charcotova nemoc, anglicky motor neuron disease MND) je onemocnění u kterého je neznámá příčina. Dochází při něm k degeneraci centrálního i periferního motoneuronu. Jde o postižení multifokální s rychlou progresí, bez remisí. Postihuje svalové skupiny končetin, trupu a bulbárního svalstva. Projevy a příznaky (tedy klinický obraz), stejně jako léčba a prognóza jsou témata, která jsou obsažena v tomto článku.
Lou Gehrigova choroba (amyotrofická laterální skleróza ALS, též Charcotova nemoc, anglicky motor neuron disease MND) je onemocnění u kterého je neznámá příčina. Dochází při něm k degeneraci centrálního i periferního motoneuronu. Jde o postižení multifokální s rychlou progresí, bez remisí. Postihuje svalové skupiny končetin, trupu a bulbárního svalstva. Projevy a příznaky (tedy klinický obraz), stejně jako léčba a prognóza jsou témata, která jsou obsažena v tomto článku.
Charcotova nemoc
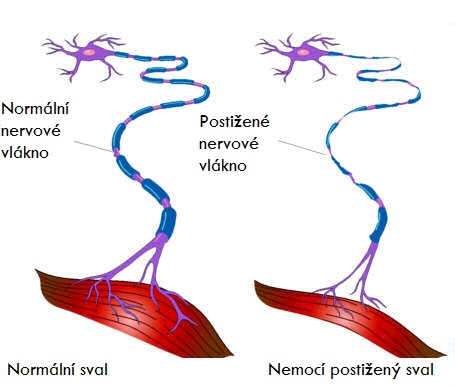
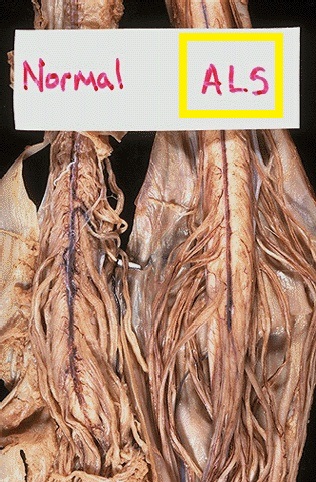
Jak již bylo psáno výše, jedná se o degenerativní onemocnění, jehož základem je rozpad motoneuronů kortexu a předních rohů míšních a to následně vede k degeneraci kortikospinálních a kortikobulbárních drah. Jde o onemocnění společensky i medicínsky velmi závažné, které je vždy letální.
Epidemiologie, incidence, etiologie (příčina) a etiopageneze (vývoj) nemoci
Muži onemocní až dvakrát častěji než ženy, incidence stoupá s věkem, maximální incidence je v dekádě 65-74 let (Dr. H. Krejčová v učebnici Speciální neurologie uvádí věk nižší a to 45-55. rok života), kdy u mužů dosahuje 10,2 a u žen 7,4 nových případů na 100 000 obyvatel/rok. Existují i familiární formy.
Etiologicky se nevýznamně uplatňují genetické faktory, povětšinou je příčina idiopatická (neznámá), v minulosti byla zvažována virová etiologie(spoluúčast retroviru), která však nebyla potvrzena (viz dále), autoimunitní mechanismus a zejména toxické působení glutatmátu (viz dále) nebo volných radikálů.
- V cytopatologickém obrazu nemoci byla opakovaně prokázána časně zduřelá neurofilamenta proximální části nervových axonů, která jsou buď primárně nebo sekundárně abnormálně fosforylována. Obsahují bílkovinu ubiquitin. V neurofibrilách a inkluzích v neuronech byly u ALS prokázány protilátky proti ubiquitinu. Zmnožení ubiquitinu však nalézáme také u Parkinsonovnemoci i u jiných degenerativních onemoěcnění.
- V motoneuronech nemocných s ALS byl prokázán edém stomatu s fragmentací endoplazmatického rtikula, chromatolýzou a rozpadem Nisslovy substance. Kromě této cytoskleletární hypotézy se zvažují i další etiologické faktory. Virová etiologie se nepotvrdila, v posleních letech je opakovaně diskutována otázka autoimunity (protilátky proti Ca a Na mbmbránovýcm kanálů) a porucha nukleových kyselin. Autoimunitní etiologie je velmi pravdoěpodobná u onemocnění, kterému říkáme paraneoplastický ALS syndrom, u kterých lze prokázat tento typ protilátek, mají vyšší pravděpodobnost současného maligního onemocnění.
- Studují se excitotoxické aminokyseliny, zejména receptory a transport neurotransmiteru glutamátu. Jeho dlouhodobé zvýšení vede svým toxickým vlivem k zániku motoneuronů. Suspektní je zejména defekt glutamátu LGT-1, kterého je v motorickém kortexu a míše u ALS nápadný úbytek. Příčinou vadného transportu glutamátu mohou být i volné kyslíkové radikály, není však jasno, zda porucha je primární nebo sekundární.
Jak se projevuje amyotrofická laterální skleróza - příznaky, projevy, symptomy
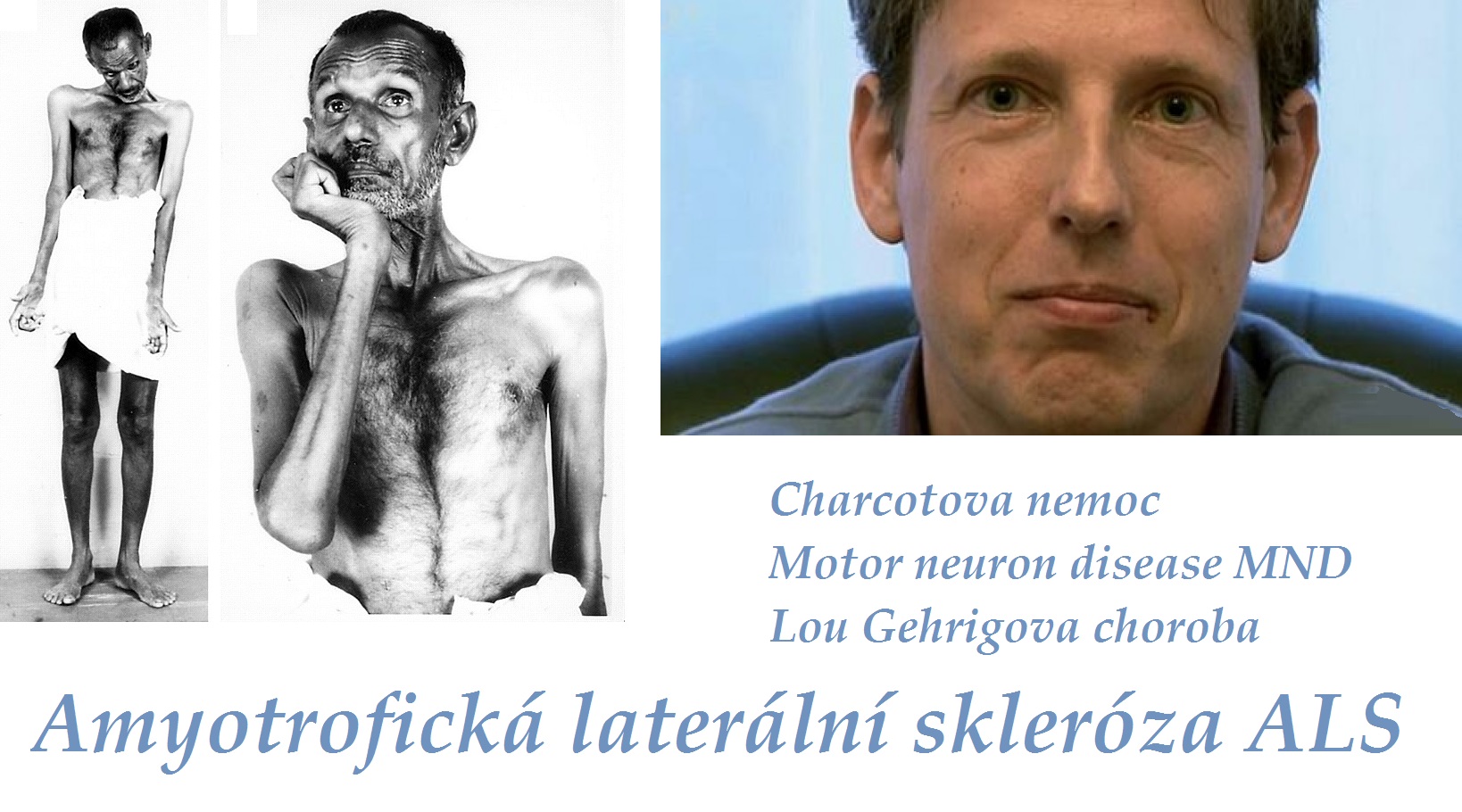
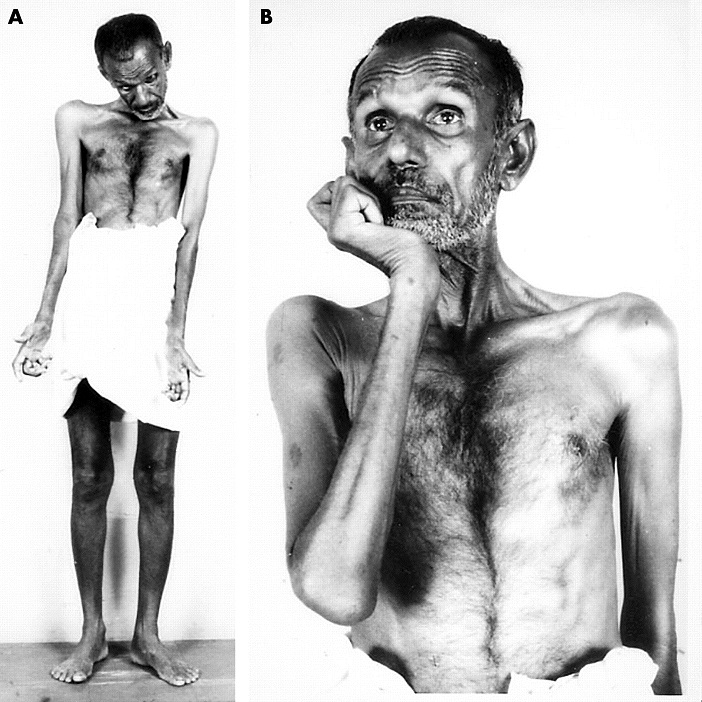
Vidíme kombinaci příznaků degenerace centrálního i periferního motoneuronu. K centrálním příznakům patří elasticita, hyperreflexie (u některých forem není podmínkou), pyramidové jevy (iritační a zánikové), klonus, bolestivé spazmy, poruchy koordinace pohybů. K příznakům periferním pak svalové atrofie, parézy, fascikulace.
Postižení má typickou triádu: atrofii svalů rukou a předloktí, spasticitu horních i dolních končetin, generalizovanou hyperreflexii bez poruchy citlivosti. U jedné třetiny nemocných začíná onemocnění na dolních končetinách (spastická paraparéza). Oslabuje se dorzální flexe nohy a prstů, objevuje se peroneální typ chůze.
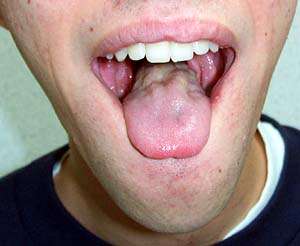
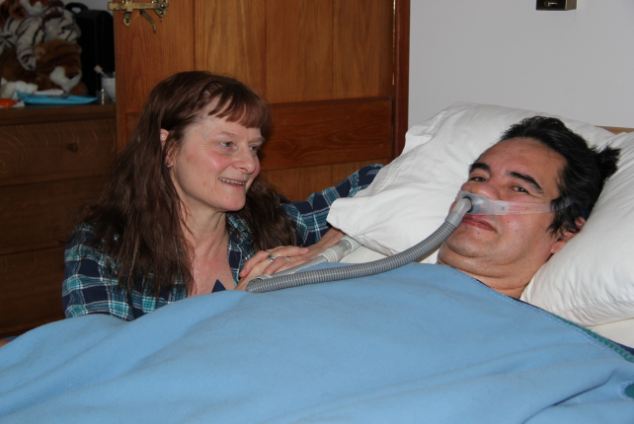
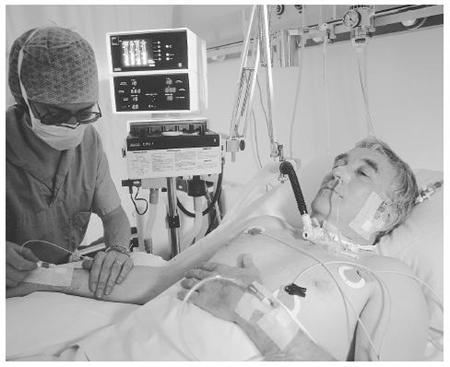
V případě bulbární formy se objeví dysartrie, porucha fonace, porucha polykání, regurgitace potravy nosem, je nebezpečí aspirace, dochází k paréze jazyka (ve vyšších stádiích ho postižený ani nevyplazí) s nepravidelnými atrofiemi a fascikulacemi. Je vyhlaslý reflex dávivý - nejprve vázne plykání tekutin a nemocný se zakuckává. Je vyšší reflex maseterový. Při paréze žvýkacích a mimických svalů bývají ústa pootevřena, z úst vytékají sliny. V této fázi je nutná výživa nazogastrickou nebo jinou sondou (nejč. perkutánní endoskopická gastrostomie PEG).
Pro všechny formy je charakteristická paréza respiračních svalů včetně bránice. Onemocnění není provázeno žádnými bolesti, paresteziemi ani poruchami čití, nedochází k poruchám funkce sfinterů, ani k okohybným poruchám.
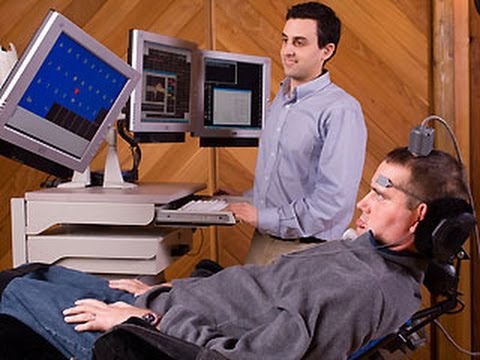
Co se týče komunikace pacienta, jeho mluva je dysartrická až anartrická, posléze vydává nesrozumitelné zvuky a dorozumívá se jen pohyby očních bulbů.
Psychika - pacient bývá motoricky neklidný, depresivní, pseudobulbární, střídavě s pláčem a inadekvátním smíchem. U více než 5% pacientů dochází k frontotemporální demenci s poruchou kognitivních funkcí a změnami afektivity (do jisté výhody se tato demence dá brát jako výhoda a vysvobození z psychických útrap).
Diagnostika
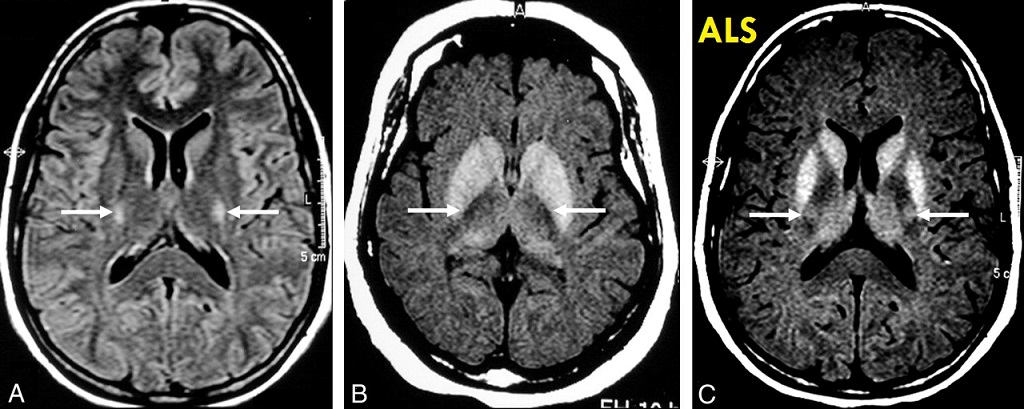
Diagnosticky přínosné je pouze EMG vyšetření (kde jsou projevy ztráty motorických jednotek, denervační projevy, vysokovoltážní potenciály motorických jednotek).
Výsledky všech ostatních vyšetření (zobrazení, likvor, ad.) jsou negativní.
Diferenciální diagnostika
Může jít i o tumory krční intumescence, cervikální myelopatii, chronickou zánětlivou polynuritidu, multifokální demyelinizující polyneuropatii a amyotrofické syndromy u malignit.
Prognóza ALS
Je vždy infaustní (smrtná). Průměrná doba přežití je 3,5 roku od stanovení diagnózy (může být ale i jen 1 rok nebo velmi vzácně i 20 let). Asi 20% postižených žije déle než 5 let, asi 10% dokonce 10let. Nemocní umírají na respirační selhání (o to hůře, že bez umělé plicní ventilace za plného vědomí!), aspirační pneumonii, embolizaci plicnice. Tracheostomie může prodloužit dobu přežití i při plegii (ochrnutí) všech kosterních svalů, kdy je zachována pouze okulomotorika (hybnost očních bulbů), kvalita života je této fázi minimální.
Varianty amyotrofické laterální sklerózy ALS
- progresivní bulbární paralýza
- progresivní spinální svalová atrofie
- primární laterální skleróza
- západopacifická forma ALS
- atypická forma Motor neuron disease s oftalmoplegií a extrapyramidovými projevy
- monomelická amyotrofie
Léčba ALS
K léčbě se používá riluzol (preparát Rilutek), který má neuroprotektivní účinek na motoneurony (potlačuje uvolňování glutamátu a bokuje glutamátové receptory). Jeho efekt je však sporný a omezený zejména na počáteční stádia nemoci. Multicentrické studie ukazují, že jeho podání prodlouží život pacienta maximálně o 6 měsíců. K imunosupresi byl využit dialyzát leukocytů. Dobrý efekt mají vazoaktiva a vitamíny skupiny B a E.
Ostatní léčba je jen paliativní (udržovací), vhodná je rehabilitace. Péče o pacienta zahrnuje sledování neurologem, logopedem, rehabilitačním lékařem, sociálním pracovníkem, plicním a ORL lékařem dle aktuálního stavu nemocného.
Související odkaz:
Amyotrofická laterální skleróza - obrázek, fotografie



Zdroje:
základ článku z medicabaze.cz, volně upraveno a doplněno dle učebnic neurologie (S. Nevšímalová - Neurologie; P. Jedlička - Speciální neurologie) a částečně z webu http://amyotroficka-lateralni-skleroza.cz/







